Some notes on the career in science of Richard F. W. Bader of McMaster University
The most important thing is to possess a passion for your subject and a willingness to work hard at it, although it never seems like work. I am always amazed that I was paid to do what I love to do and think about. Everything else will follow. You never consider any possible course other than the one that will enable you to pursue your passion. My family was not "well-off" and I was to be the first in my immediate family to attend university. I had applied to McMaster University for a regional scholarship (I was born (in 1931) and bred in Kitchener), because without it, the financial constraints would have been considerable. I spent the summer after completing grade 13 pulling toe nails off of the front hooves of pigs, the first station in an assembly line after they had been slaughtered and hung up on struts. My father worked in the same factory and one afternoon in August he came to me on the "killing floor" to tell me that I had been awarded the scholarship to McMaster. I said "great, now I can quit this job." But of course, he did not let me quit. Among the most important things my father taught me was never to quit. This is a most important lesson in science. Working on new ideas and new approaches in science or art - or any human endeavor, places one at odds with those content with the prevailing ideas or mores - the existing paradigms. One must believe in what one is doing and stay the course. Now of course, in science one can ultimately prove the correctness of one's work by appeal to experiment and established theory. But even with this buttressing of one's ideas, acceptance can be a long and difficult road.
The goal of a scientist is to uncover new ideas, concepts and tools, practical or theoretical, that extend our understanding of the world around us and enable us to do new things. In chemistry, the theory of Atoms In Molecules (AIM) developed in my laboratory, is being increasingly used by workers in all fields of chemistry and also in solid sate physics. Because AIM has increased our understanding of how atoms behave, it is used in many ways - to develop new alloys and new and better drugs, for example. Nothing pleases me more than the knowledge that each year sees an ever increasing use of AIM by other scientists in the pursuit of their own research.
My earliest recollections are of the interest I had in observing things in my surroundings. My father kept a large garden, both flowers and vegetables, and I spent good deal of time there studying not only the plants but the attendant insects and birds. Perhaps "studying" is too pretentious a word. I kept no notes but I recall performing crude experiments, such as attempting to mend a broken stalk of a cucumber plant. I still have microscope slides I mounted of plant and animal tissue from those days, one slide being labeled "Murray's blood", Murray being a boyhood chum. This was accompanied by star gazing and learning to identify the major constellations. Then one day I learned about chemistry and my life changed forever. I very quickly assembled a good laboratory (not one from a chemistry set) and kept copious notes on the experiments I performed. I became known to my friends as the "mad scientist" and I entertained them occasionally with small explosions. One day I recall quite vividly: I was generating hydrogen sulphide gas using a Kipp generator when something went terribly wrong and the gas began to escape into the room. My "lab" was in the basement of our house, next to the furnace and the noxious gas quickly spread throughout the house via the furnace pipes. My father was upstairs in his study trading stamps with an elderly gentleman who promptly became quite ill. After my father led the gentleman out of doors, I heard him descending the basement stairs, causing me to panic and flee the scene. My father grabbed the Kipp generator and threw it out the basement window, saving the day. He was after all a Scout master, and so knew how to deal with emergencies.
I have never been a fan of lectures or articles that purport to show young people that science or chemistry can be "fun" in attempts to lure them into a scientific career. That you are going to devote you life to science is something you just know. One aspect of science that appealed greatly to me by the time I was in my early teens, was that science is without prejudices - that truth, as acquired by observation, would always prevail. One builds upon the work of others that have preceded you, but then you are the master of your own fate, free to work on what you wish, as long as you follow the rules of the game - experiment and observation followed by theory. If you are fortunate enough to initiate something new, you must be prepared to have others telling you that you are wasting your time, as I was frequently told after I decided in 1959 that the vehicle for understanding chemistry was the electron density. The electron density is now, of course, routinely measured and used not only by those who apply the theory of atoms in molecules in predicting and understanding the properties of matter, but it also forms the basis for density functional theory that has greatly extended the range of applicability of computational chemistry and physics.
Something that helped and guided me in my search for a theoretical understanding of the concepts of chemistry was my strong grounding in experimental chemistry. I began my professional life as an experimental chemist taking a Master's degree in physical organic chemistry under Professor A. N. Bourns at McMaster University, a field I continued to study for my PhD studies at the Massachusetts Institute of Technology (MIT) under Professor C. G. Swain. My studies concerned reaction mechanisms and it became clear from the many discussions - and arguments - I had with Professor Swain that chemists operate under a considerable handicap. Chemists use the notions of atoms, bonds and structure but everyone has their own understanding of these terms. How could we discuss the making and breking of bonds between atoms when we had differing ideas of what constituted an atom and a bond? Chemists possessed a common language, but everyone had their personal dictionary. It was after completing my degree at MIT in 1958 that I left for Cambridge University to study theoretical chemistry under Professor Longuet-Higgins with the express purpose of developing a theoretical understanding of the language of chemistry with the goal of providing everyone with a common dictionary.
When I arrived at Cambridge I found a series of papers by Professor A. C. Hurley who had been a visitor at the laboratory, before my arrival, and then under the tutelage of Professor Lennard-Jones. They offered a beautiful, clear and physical understanding of chemical binding in terms of the forces exerted on the nuclei by the electron density, as determined by the Hellmann-Feynman theorem (HFT). The HFT shows that the forces exerted on the nuclei in a molecule are understood and calculable using classical electrostatics, with the force of attraction being determined by the electron density. Once quantum mechanics is used to determine the density of electrons, then the discussion of the forces experienced by the nuclei is rigorously couched in the language of classical electrostatics. Better still, one deals with the real, space-filling, measurable electron density rather than the illusory orbitals that existed only in some mathematical space. I read as well the paper by Berlin wherein he defined the binding and antibinding regions of a diatomic molecule and showed how binding was a result of the accumulation of sufficient electron density in the binding region between the nuclei. I never looked back and after my return to Canada, eventually to McMaster University, my research interests centred on the electron density.
Observational Basis For The Theory Of Atoms In Molecules
I was most fortunate in this endeavor in forming a collaboration with Professor Mulliken's laboratory for molecular structure and spectra (LMSS) at the University of Chicago. His group, along with Professor Roothaan, were in the process of obtaining wave functions of near Hartree-Fock quality for hundreds of diatomic molecules; both homonuclear and heteronucler, in ground and in excited states. I was given access to these functions, working directly with Dr. P. E. Cade of LMSS in the late 60s and early 70s, work that resulted in the first comprehensive study of the properties of molecular electron density distributions, published as a series of papers in the Journal of Chemical Physics. Electron densities of crystals are now routinely obtained from X-ray scattering studies, but in our work, the LMSS wave functions were the source of the densities on which we made our "experimental observations". These studies led to the realization in 1972 that the topology of the electron density (hereafter labeled  (r)) provided a unique and "natural" partitioning of the space of a molecule or a crystal into mononuclear regions that I shall refer to as atoms. Equally important, it soon became apparent that the resulting regions were transferable to varying extents between molecules. Indeed, if one insisted that the pieces transferred had to exhaust the system, they had the property of maximizing any transferability that was present.
(r)) provided a unique and "natural" partitioning of the space of a molecule or a crystal into mononuclear regions that I shall refer to as atoms. Equally important, it soon became apparent that the resulting regions were transferable to varying extents between molecules. Indeed, if one insisted that the pieces transferred had to exhaust the system, they had the property of maximizing any transferability that was present.
The natural partitioning of space, as described in detail below, is accomplished by the "zero-flux surface", a surface determined by the topology of
(r). Here I wish to emphasise the initial excitement that was incurred by the realisation that the mono-nuclear regions so defined recover the chemistry of an atom in a molecule, as described in J. Chem. Phys. 56, 3320 (1972). This natural definition satisfies the two essential requirements imposed on any possible definition of an atom  in a molecule that stem from the concept of a functional group: (1) it maximises the transferability of the atom's distribution of charge from one system to another and (2) this transferability in the density is accompanied by a paralleling transferability in the other properties of the atom including its energy. This finding is but the satisfaction of the dictum that two atoms that look the same must necessarily make the same contributions to all properties. Point (2) was a result of parallel studies that we were conducting on the kinetic energy density of the electrons, a function that, like the electron density
(r), is defined in real space. (It should also be pointed out that the zero-flux partitioning method yields a unique definition of T(),
the electronic kinetic energy of the atom.) We made what was to be the most important of all our observations - that the transferability of the kinetic energy density parallels the transferability of
(r). Two atoms that "looked" the same in real space would posses the same electronic kinetic energy. Amazing, because just suppose the virial theorem of quantum mechanics applied to such a topological atom, then one could define the total energy of atom in a molecule! This follows from the statement of the virial theorem that equates the total energy E to the negative of the kinetic energy T, for a system governed by Coulombic forces. For an atom in a molecule, one could equate E() to - T(), where E(
) is the total electronic energy and T(
) its electronic kinetic energy. The same identification enables one to relate the total energy of the molecule E to the sum of the atomic contributions, that is,
in a molecule that stem from the concept of a functional group: (1) it maximises the transferability of the atom's distribution of charge from one system to another and (2) this transferability in the density is accompanied by a paralleling transferability in the other properties of the atom including its energy. This finding is but the satisfaction of the dictum that two atoms that look the same must necessarily make the same contributions to all properties. Point (2) was a result of parallel studies that we were conducting on the kinetic energy density of the electrons, a function that, like the electron density
(r), is defined in real space. (It should also be pointed out that the zero-flux partitioning method yields a unique definition of T(),
the electronic kinetic energy of the atom.) We made what was to be the most important of all our observations - that the transferability of the kinetic energy density parallels the transferability of
(r). Two atoms that "looked" the same in real space would posses the same electronic kinetic energy. Amazing, because just suppose the virial theorem of quantum mechanics applied to such a topological atom, then one could define the total energy of atom in a molecule! This follows from the statement of the virial theorem that equates the total energy E to the negative of the kinetic energy T, for a system governed by Coulombic forces. For an atom in a molecule, one could equate E() to - T(), where E(
) is the total electronic energy and T(
) its electronic kinetic energy. The same identification enables one to relate the total energy of the molecule E to the sum of the atomic contributions, that is,  . We had in our hands a definition of an atom in a molecule that accounted for the obvious requirements that two atoms that look the same, possess the same energy and that when summed over all the atoms, yielded the total energy.
. We had in our hands a definition of an atom in a molecule that accounted for the obvious requirements that two atoms that look the same, possess the same energy and that when summed over all the atoms, yielded the total energy.
In the year following, the full topological significance of the zero flux surface condition was realised, leading to a complete theory of molecular structure and structural stability. (J. Chem. Phys. 70, 4316 (1979)). A brief review of the principal topological features of the theory is given in the following paragraphs. A full account is given in "A topological theory of molecular structure", Reports on Progress in Physics, 44, 893-948 (1981).
An Introduction To The Topology Of The Electron Density: The Definition Of An Atom
Matter is made manifest through the space-filling distribution of charge. A charge distribution consists of point-like nuclei embedded in a diffuse cloud of negative charge whose spatial distribution is described by the electron density,
(r). The topology of the electron density is dominated by the attractive force exerted on it by the nuclei, a force that endows the density with its principal topological feature - that it exhibits a maximum value at the position of each nucleus, a feature that is true for any plane containing a nucleus, as displayed for two planes of the molecule BF3, Fig. 1(a) and (b). An immediate consequence of this topological feature of the density is the natural association of an atom with a region of space, each region being dominated by a given nucleus, with boundaries evident in the minima that exist between the nuclear maxima. The boundaries are determined by the balance in the forces that the neighbouring nuclei exert on the density.
The definition of an atom and its surface are made both qualitatively and quantitatively apparent in terms of the patterns of trajectories traced out by the gradient vectors of the density, vectors that point in the direction of increasing
. Trajectory maps, complementary to the displays of the density, are given in Fig. 1(c) and (d). Because
exhibits a maximum at each nucleus in any plane that contains the nucleus (the nucleus acts as a global attractor), the three-dimensional space of the molecule is divided into atomic basins, each basin being defined by the set of trajectories that terminate at a given nucleus. An atom is defined as the union of a nucleus and its associated basin. The saddle-like minimum that appears in the planar displays of the density between the maxima for a pair of neighbouring nuclei is a consequence of a particular kind of critical point (CP), a point where all three derivatives of
vanish, that is, where 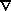 = 0. The density is a minimum at this CP along a line linking the two nuclei, a line that is uniquely defined by the pair of trajectories that originate at this CP and terminate at the two adjoining nuclear maxima, as indicated by a pair of arrows on the trajectory plot for one B-F interaction in Fig. 2. The line so defined represents a line of maximum density with respect to any neighbouring line and for a bound molecule, it is referred to as a bond path, and the associated CP a bond critical point. A bond path provides a universal indicator of bonding, linking all pairs of bonded atoms, regardless of the nature of the interaction. A molecular graph, defined by a molecule's connected set of bond paths, defines a molecular structure with particular critical points defining the presence of bonded rings and cages. Nuclear motions can induce topological changes in the density that correspond to the making and breaking of chemical bonds and to a change in molecular structure. Thus the topology of the charge distribution and its change, as induced by nuclear motions, embodies the essential elements of structure and structural change.
= 0. The density is a minimum at this CP along a line linking the two nuclei, a line that is uniquely defined by the pair of trajectories that originate at this CP and terminate at the two adjoining nuclear maxima, as indicated by a pair of arrows on the trajectory plot for one B-F interaction in Fig. 2. The line so defined represents a line of maximum density with respect to any neighbouring line and for a bound molecule, it is referred to as a bond path, and the associated CP a bond critical point. A bond path provides a universal indicator of bonding, linking all pairs of bonded atoms, regardless of the nature of the interaction. A molecular graph, defined by a molecule's connected set of bond paths, defines a molecular structure with particular critical points defining the presence of bonded rings and cages. Nuclear motions can induce topological changes in the density that correspond to the making and breaking of chemical bonds and to a change in molecular structure. Thus the topology of the charge distribution and its change, as induced by nuclear motions, embodies the essential elements of structure and structural change.
The density is a maximum in all directions perpendicular to the bond path at the position of a bond CP, and it thus serves as the terminus for an infinite set of trajectories, as illustrated by arrows for the pair of such trajectories that lie in the symmetry plane shown in Fig. 2. The set of trajectories that terminate at a bond critical point define the interatomic surface that separates the basins of the neighbouring atoms. Since the surface is defined by trajectories of
that terminate at a point, and since trajectories never cross, an interatomic surface is endowed with the property of zero-flux - a surface that is not crossed by any trajectories of
, a property made clear in Fig. 2 and expressed in the equation
 (1)
(1)
The final set of diagrams in Fig. 1 display contour maps of the electron density overlaid with trajectories that define the interatomic surfaces and the bond paths to obtain a display of the atomic boundaries and the molecular structure.
Laplacian of the Electron Density and the Lewis Model
The topology of the density, while recovering the concepts of atoms, bonds and structure, gives no indication of the localised bonded and nonbonded pairs of electrons of the Lewis model of structure and reactivity, a model secondary in importance only to the atomic model. The Lewis model is concerned with the pairing of electrons, information contained in the electron pair density and not in the density itself. Remarkably enough however, the essential information regarding the spatial pairing of electrons is contained in the Laplacian of the electron density, the sum of the three second derivatives of the density at each point in space, the quantity
2
(r).
It is a property of the second derivative of a scalar function such as
that it determines where the function is locally concentrated and locally depleted, in the absence of corresponding maxima and minima in the function itself:
2
(r) < 0 implies that
(r) is greater at r than the average of its values in the immediate neighbourhood of r, with the reverse being true for
2
(r) > 0. It is useful to define the function L(r) = -
2
(r), since a maximum in L(r) then corresponds to a maximum in the concentration of electronic charge.
The topology of L(r) is completely different from that for the density itself, exhibiting local maxima corresponding to the presence of Lewis bonded and nonbonded electron pairs. It has been known for some time that L(r) recovers the shell structure of an atom in terms of a corresponding number of alternating pairs of shells of charge concentration and charge depletion and that upon bonding with other atoms, the outer or valence shell of charge concentration loses its uniformity resulting in the formation of local maxima, that is, local charge concentrations (CCs). The number, relative size and orientation of these CCs provide a faithful mapping of the localised bonded and nonbonded Lewis pairs assumed in the VSEPR model of molecular geometry. Indeed, it has been demonstrated that the topology of L(r) provides a mapping of the essential information of the conditional pair density, from six- to three-dimensional space and the mapping of the topology of L(r) onto the Lewis and VSEPR models is grounded in the physics of the pair density. Readers are encouraged to read the 1916 paper by G. N. Lewis (if they have not already done so) wherein he introduced the concept of the electron pair and its role in chemistry - before the advent of quantum mechanics! (J. Am. Chem. Soc. 38, 762 (1916)).
Further Reading
My book, "Atoms in Molecules: A Quantum Theory" published in 1990 by the University of Oxford Press, contains an introduction to both the topological and quantum mechanical aspects of the theory. A summary of the theory is given in the article "Why are there atoms in chemistry?", Can. J. Chem. 76, 973-988 (1998). The extension of the theory to include the effects of magnetic fields is to be found in the following papers: J. Chem. Phys. 99, 3669-3682 and 3683-3693 (1993). Reference to the recent literature will yield applications to many fields.
Figures
Figure Captions
- The electron density
(r) is displayed in the F
h and F
v symmetry planes of BF3 in (a) and (b), respectively. The density is a maximum at the position of each nucleus (values of
greater than 2.5 au are not shown in the relief maps) and displays a saddle between B and each of the F nuclei. The minimum in
at a saddle point denotes the position of a bond critical point (BCP). The trajectories traced out by the vectors
are illustrated in (c) and (d) for the same planes as in (a) and (b). All the paths in the neighbourhood of a given nucleus terminate at the maximum value in
found at each nucleus and define the atomic basin. Figs. (a) and (b) show two orthogonal views of the same BCP. They indicate that
is as minimum at the BCP along the internuclear axis, the curvature is positive, and two trajectories originate at a BCP and terminate at the two associated nuclei. They define the bond path, a line of maximum electron density linking bonded nuclei. The curvature of
is negative in every direction perpendicular to the bond path and
appears as a maximum at the BCP. An infinite set of trajectories thus terminate at the BCP and define the interatomic surface. A BCP is located at the intersection of the bond path and the interatomic surface. 1(e) and (f) show the electron density in the same two planes overlaid with the bond paths and the interatomic boundaries. The values of the contours in this and following figures, increase from the outermost 0.001 au contour inwards in steps of 2H
10n, 4H
10n and 8H
10n au with n beginning at -3 and increasing in steps of unity.
- A display of the trajectories of
for the same plane as in Fig. 1(d), complemented with arrows denoting the two unique trajectories that originate at the BCP, marked by an open circle, and terminate at each of the neighbouring nuclei. They define the bond path. Also indicated by arrows are the two trajectories that terminate at the BCP in this symmetry plane. They are members of the infinite set of such trajectories that define the interatomic surface of zero-flux in
between the boron and fluorine atoms.