| |
MICHAEL J. McGLINCHEY
CURRENT RESEARCH INTERESTS |
Home |
All of our current
projects involve the syntheses, characterization and molecular dynamics of organometallic
molecules. These systems generally exhibit interesting fluxional behaviour and so
require an investigation not merely of the nature of the rearrangements and the barriers
associated with them, but also of the underlying electronic reasons for the ease or
difficulty of the processes. Structures and dynamics are elucidated principally by
means of NMR spectroscopy, mass
spectrometry and X-ray
crystallography. Molecular orbital calculations are used to probe the nature of
the frontier orbital interactions, and all members of the group are encouraged to become
proficient in the use of each of these techniques. We have long-standing
collaborations with many other groups both at McMaster University, and elsewhere in Europe
and North America, and regularly exchange graduate students and post-doctoral research
associates in both directions.
The
following brief summaries give an overview of some of our recent and current
activities. Note that reference numbers in the text correspond to those in the publication list.
- Organometallic derivatives of natural
products: (a) hormonal steroids and receptor sites, (b) terpenes and "non-classical" cations
|
Sterically crowded organo-transition
metal complexes pose interesting problems in terms of their molecular dynamics.
For example, the observation that the
interconversion of proximal and distal ethyl groups in (C6Et6)Cr(CO)3
can be slowed on the NMR time-scale prompted us to ask whether ethyl rotation is
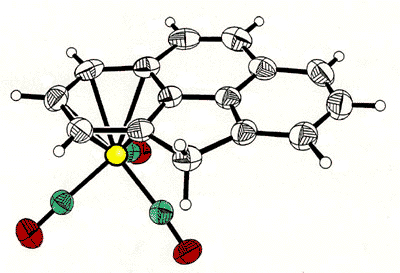
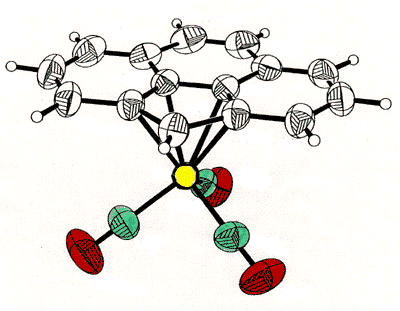
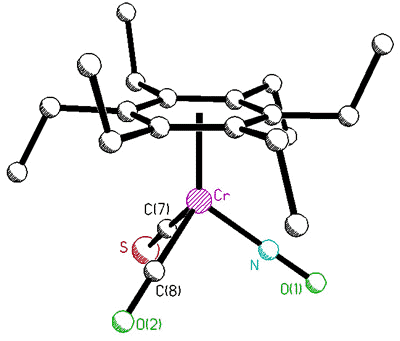
correlated with that of the tripod [63]. To probe this phenomenon, Dr. Bavani
Mailvaganam synthesized the cation [(C6Et6)Cr(CO)(CS)(NO)]+
whose variable-temperature 13C NMR spectrum exhibits at low temperature
eighteen carbon environments (6 methyl, 6 methylene, 6 ring carbons) for the
hexaethylbenzene ligand [107]. However, she found that the barriers to tripodal
rotation (9.5 kcal/mol) and ethyl rotation (11.5 kcal/mol) are different, showing that
these processes are not correlated.
 |
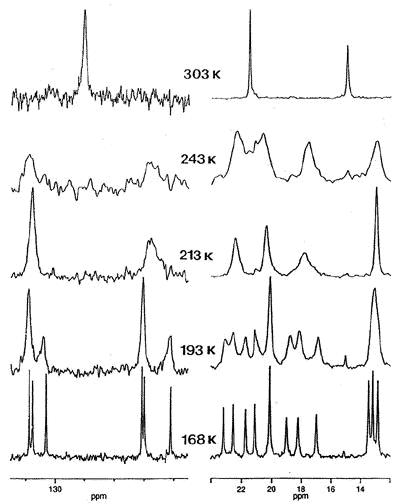 |
X-ray crystal structure, and variable-temperature 125.8 MHz 13C
NMR spectra of [(C6Et6)Cr(CO)(CS)(NO)]+ |
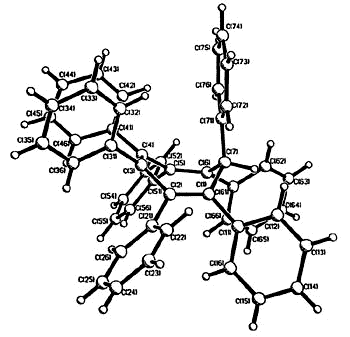
Moreover, complexes containing C5Ph5,
C6Ph6 or C7Ph7 ligands exhibit conformations
in which the polyphenylated fragments adopt chiral propeller-type structures. The
peripheral rings in these molecules adopt propeller-like structures as a compromise
between a planar, but sterically inaccessible, geometry and the completely orthogonal
orientation in which all conjugation with the central ring would be lost. Such
molecules can be locked into chiral conformations and the barriers to the interconversion
of such enantiomers (or diastereomers) can be probed by a variety of NMR techniques
[126,103,136]. Concurrent with the experimental work, computational studies are
under way in a collaborative venture with Professors
Kim Baldridge and Jay Siegel (University of California, San Diego).
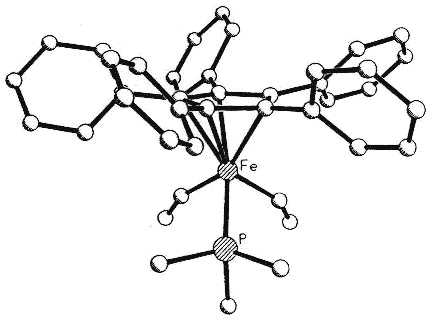 |
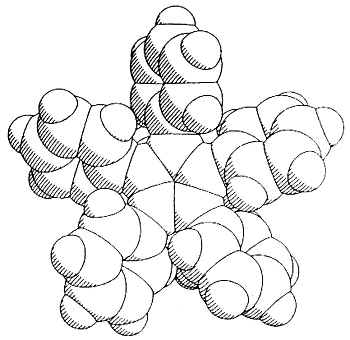 |
Side view & space-fill of (C5Ph5)Fe(CO)(CHO)PMe3
emphasising the propeller conformation of the C5Ph5 group
|
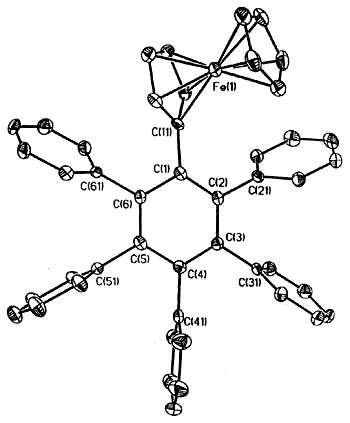
|
X-ray crystal structure of
ferrocenyl-pentaphenylbenzene
|
 |
 |
| X-ray
crystal structure of heptaphenylcycloheptatriene |
X-ray crystal structure of C7Ph6FcH |
As the ring size increases, steric
crowding between peripheral phenyl rings becomes so severe that the 6- Hückel system [C7Ph7]+
is non-planar. The cation was synthesized by Dr. Hari Gupta, and the X-ray
crystal structure was solved by Stacey Brydges [162].
Hückel system [C7Ph7]+
is non-planar. The cation was synthesized by Dr. Hari Gupta, and the X-ray
crystal structure was solved by Stacey Brydges [162].
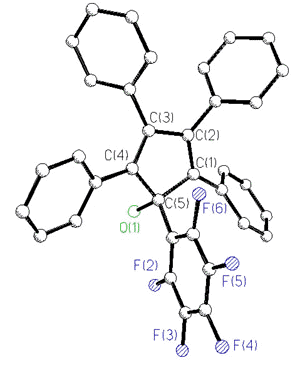
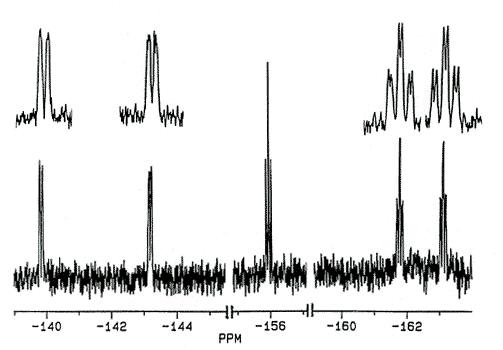
In an attempt to incorporate a probe to
monitor the barrier to peripheral phenyl rotation in cyclopentadienyl systems, the ligand
C5Ph4(C6F5)OH has recently been prepared
[170]. The 19F spectra of this and related molecules exhibit five
fluorine environments at room temperature; the pentafluorophenyl rotational barrier
exceeds 20 kcal/mol.
X-Ray crystal
structure and 282 MHz 19F NMR spectrum of C5Ph4(C6F5)OH
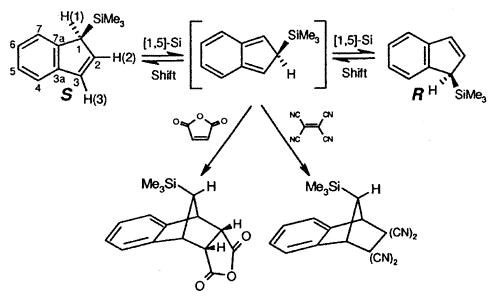
Sigmatropic migrations, such as
those shown for S and R trimethylsilylindene, interconvert stereoisomers
(enantiomers or diastereomers, depending on the molecule under investigation), and these
rearrangement mechanisms have attracted much interest. In the particular case depicted,
the process occurs via successive [1,5]-suprafacial migrations and the intermediate iso-indene
can be intercepted as a Diels-Alder adduct [150].
 |
 |
| Interconversion
of enantiomers of TMS-indene, and trapping of the intermediate iso-indene by
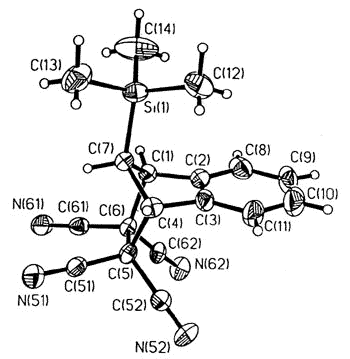
either maleic anhydride or TCNE |
X-ray crystal structure of the TCNE adduct |
The stereochemical complexity of these
systems can become relatively severe and, in the tris-indenyl-silane series, investigated
by Dr. Mark Stradiotto, the pathways for NMR site exchange can be mapped on a hypercube
[150].
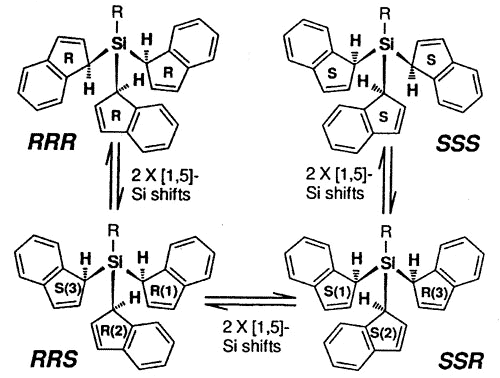
Interconversion of
the eight different indenyl ring environments in the isomers of tris(1-indenyl)silanes
In cases where the molecule is
temperature-sensitive, simple variable-temperature NMR using line-broadening techniques
may be inapplicable and more sophisticated methods, such as 2D-EXSY or single selective
inversion methods, may be required. We have an ongoing collaboration with our
colleague Professor
Alex Bain to study such systems.
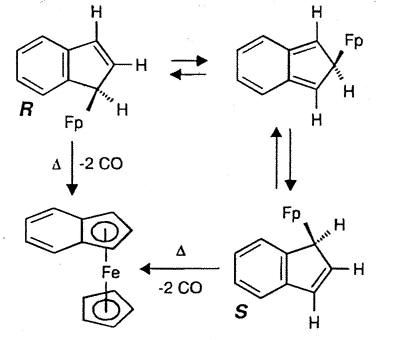
Typically, (eta-1-indenyl)Fe(CO)2(C5H5)
was for many years believed to be non-fluxional; moreover, it readily loses two carbonyl
ligands to generate benzoferrocene. However, the 2D-EXCHANGE map shown below clearly
reveals the existence of a fluxional process even at a relatively low temperature. Once
again, the intermediate iso-indene has been trapped as a TCNE cycloadduct [159].
 |
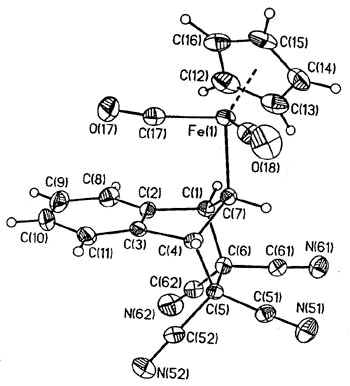 |
| Interconversion of
the enantiomers of (eta-1-indenyl)- Fe(CO)2(C5H5), and
decarbonylation to yield benzoferrocene |
X-ray
crystal structure of the iso-indene-TCNE adduct |
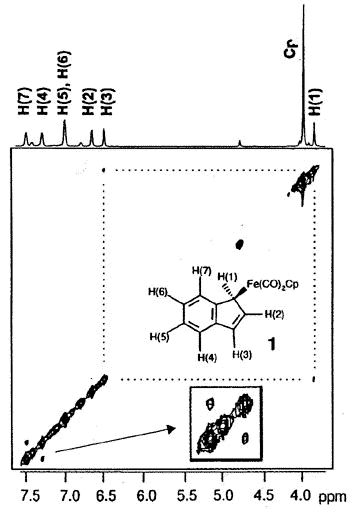
2-D EXSY spectrum of
(eta-1-indenyl)Fe(CO)2(C5H5) showing exchange between
H(1) and H(3), and between H(4) and H(7) sites
Occasionally, iso-indenes are
sufficiently stabilized, by incorporation of multiple aromatic rings, that they can react
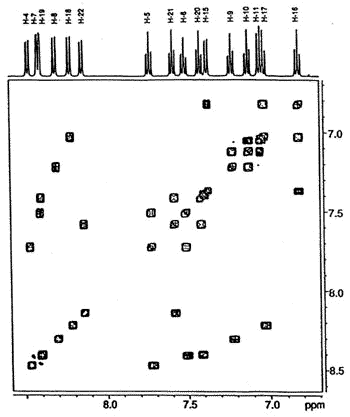
with their progenitor to yield a Diels-Alder adduct. Typically, Dr. Suzie Rigby
showed that cyclopenta[l]phenanthrene and its corresponding iso-indene give
rise to the dimer (illustrated below) that has been characterized by NMR spectroscopy and
X-ray crystallography [161].
![cylopenta[l]phenanthrene dimer](images/suziedimer.gif) |
|
| Diels-Alder
dimerization of cyclopenta[l]phenanthrene with its own iso-indene isomer |
500 MHz 1H-1H
COSY NMR spectrum of the dimer showing the connectivity between the protons in the four
different aromatic rings
|
![cyclopenta[l]phenanthrene dimer xray](images/suzie92.gif) |
![cyclopenta[l]phenanthrene dimer xray](images/suzie91.gif) |
X-ray crystal structure of the Diels-Alder adduct,
emphasising the endo nature of the dimer |
For more examples of silatropic shifts in
polycyclic systems, see references 139, 143, 148, 160, 167 and 171.
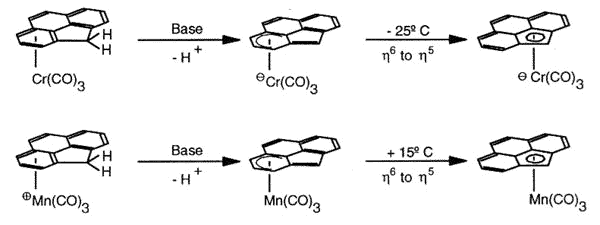
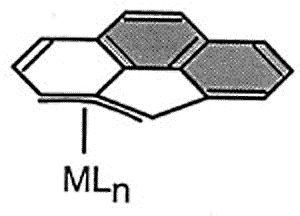
Haptotropic shifts involve the
migration of an organometallic fragment (such as Mn(CO)3) between rings, as
exemplified by the interconversion of the eta-6 and eta-5 isomers of (cyclopenta[def]phenanthrenyl)MLn,
which represents 25% of the C60 framework. Crystallographically characterized
molecules depicting "before" and "after" situations are shown below
[122]. This work is taken from the thesis of Dr. Andreas Decken.
As previously discussed for indenyl,
naphthalene and related systems (Albright, T.A.; Hofmann, P.; Hoffmann, R.; Lillya, C.P.;
Dobosh, P.A. J.Am. Chem. Soc. 1983, 105, 3396), the organometallic
moiety does not execute a "least-motion" trajectory across the common bond
between the rings, but instead follows a rather circuitous pathway along the molecular
periphery so as to optimise the aromatic character of the transition state.
|
|
The diagram shows the most favorable pathway for a
manganese tricarbonyl moiety undergoing a haptotropic shift across a cyclopenta[def]phenanthrenyl
ligand from a 6-membered ring to the 5-membered ring; note how the orientation of the
tripod changes during the migration process. |
The exocyclic trajectory followed
by the organometallic fragment is favored by the development of a 'naphthalene-type"
aromatic transition state.
|
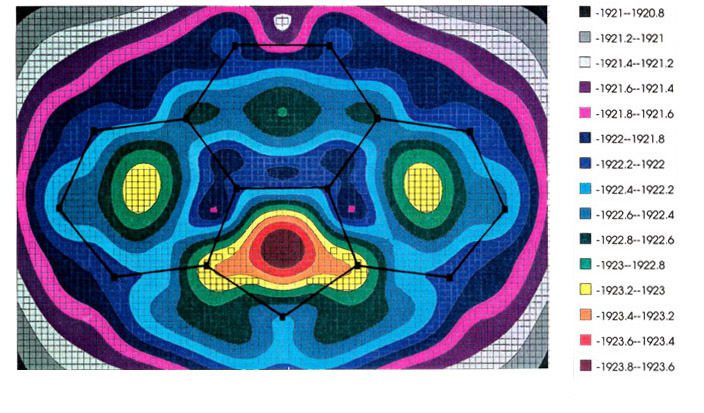
The most favored trajectory can be
elucidated by use of molecular orbital calculations to generate an energy hypersurface
whereby the organometallic fragment is systematically moved over the ligand framework so
as to find the local minima, and the pathways that connect them. The examples shown
illustrate the migration of (a) a manganese tricarbonyl fragment and (b) a
cyclopentadienyl-iron unit over the surface of a cyclopenta[def]phenanthrenyl
ligand; energy minima are indicated by increasing red coloration, while blue and purple
represent regions of relative instability [151]. In both the Mn and Fe cases (upper
and lower pictures, respectively), note the high barrier where the organometallic fragment
crosses the common bond between the six- and five-membered rings. [The energy scale is in
eV.] These calculations were carried out by using the program CACAO,
written by Dr.
Carlo Mealli.

Current work is aimed at synthesizing
metal complexes of non-planar polycyclics, e.g. sumanene, C21H12,
that are fragments of fullerenes, and elucidating their haptotropic behaviour [154,155].
Transition Metal Stabilized Reaction
Intermediates
The use of organometallic moieties or
metal clusters to allow the isolation and characterization of short-lived species not only
yields structural data on otherwise unavailable systems, but also provides a method of
storing these reactive intermediates until required for synthetic purposes.
Benzylic anions and cations are readily
generated as chromium tricarbonyl complexes and their geometries and molecular dynamics
have been investigated both experimentally (by NMR) and theoretically [58,118].
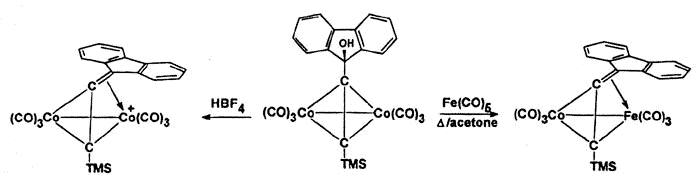
Propargylic cations are particularly well stabilized as dicobalt hexacarbonyl clusters
(Nicholas, K.M. Acc. Chem. Res. 1986, 20, 207). Although they are
valuable synthetic intermediates, and have been intensively investigated by a variety of
spectroscopic and theoretical methods, crystallographic characterization remains a serious
challenge (Melikyan et al. Angew. Chem, Int Ed. Engl. 1998, 37, 161).
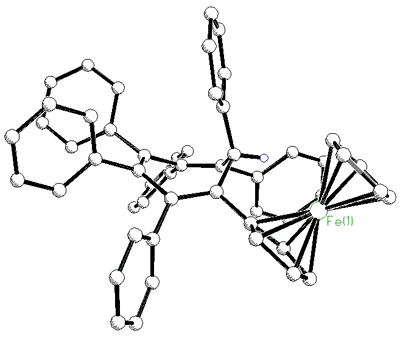
However, isolobal replacement of a Co(CO)3+ vertex by a neutral
Fe(CO)3 group [124], as in the fluorenyl cluster depicted below (synthesized
and crystallographically characterized by Dr. James Dunn), has proven an excellent method
of modelling the structures of the analogous cobalt-stabilized cations [168].
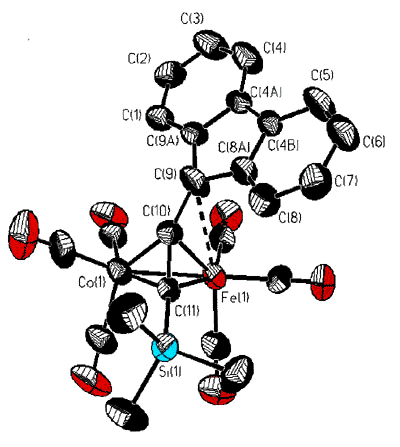
X-ray crystal
structure of the mixed iron-cobalt fluorenyl cluster
A related project, in collaboration with
our colleague Professor
M.A. Brook [132,156,164] involves the development of metal cluster stabilized silicon
cations, which are currently isolable only when partnered by counter-ions of exceptionally
low nucleophilicity.
Sometimes, serendipity plays a role, as
when Dr. Luc Girard treated 1,2,4,5 tetra(bromomethyl)benzene with Na2Fe(CO)4
and produced the first metal complex of the long-sought disjoint diradical
tetramethylenebenzene [129].
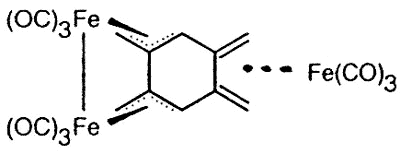 |
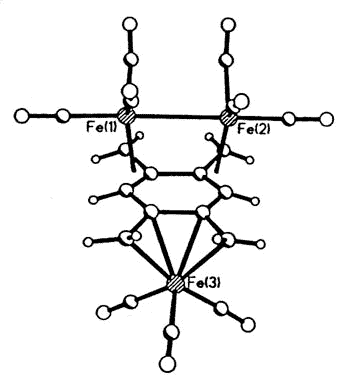
|
X-ray crystal structure of
(1,2,4,5-tetramethylenebenzene)tris-irontricarbonyl |
Organometallic Derivatives of Natural
Products:
(a) Metal Complexes of Hormonal Steroids
Complexation of organometallic fragments
to steroids serves many purposes: the placement of an Fe(CO)3 unit on the
alpha-face of ergosteryl acetate (or dehydrocholesteryl acetate, shown below), protects
the B-ring from attack and permits functionalization of the side-chain. The X-ray
crystal structure was solved by Dr. Richard Perrier [115].
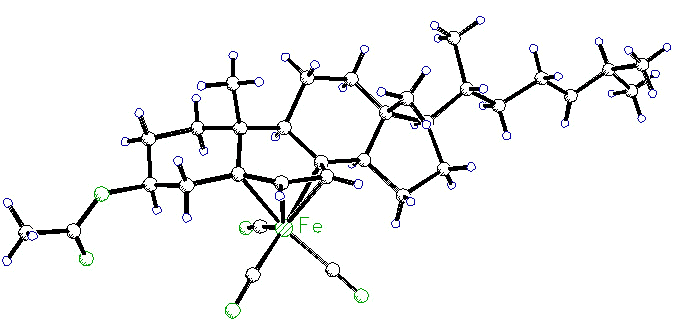
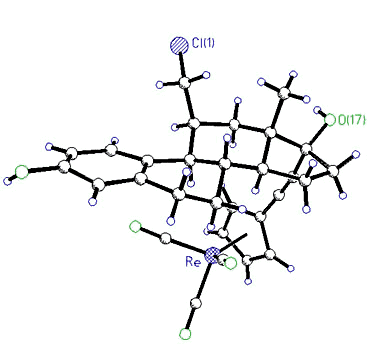
More recently, hormonal steroids have
been synthesized that bear ruthenium [98] or rhenium [114,140] moieties and have a
high affinity for estradiol receptor sites. The longer term goal is to incorporate
appropriate radio-isotopes (e.g. 106Ru, 186Re, 188Re)
with which to image or eradicate cancerous tumours. The structures of two typical
molecules are shown below:
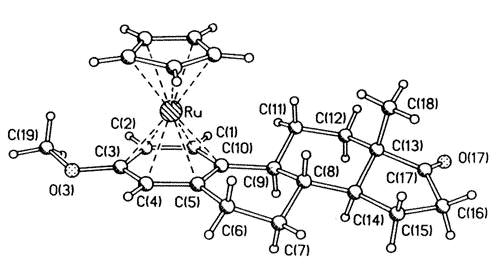 |
 |
| X-ray
crystal structure of the ß-[CpRu(estrone 3-methyl ether)] cation |
X-ray
crystal structure of 11ß-(chloromethyl)-17 -[(CC-C5H4)Re(CO)3]-estradiol -[(CC-C5H4)Re(CO)3]-estradiol |
This is a collaborative project with
Professor Gérard Jaouen and Dr. Anne Vessičres in Paris. More information is available
on the website of their laboratory, the Laboratoire de Chimie
Organométallique, ENSCP, Paris.
(b) Terpenes and
"Non-classical" Cations.
Wagner-Meerwein rearrangements of
terpenes have held a particular fascination for chemists for more than a hundred years.
Indeed, the question of "classical" versus "non-classical"
carbocations has been an exceedingly contentious topic. In light of the known ability of
transition metal clusters to stabilize cations, we chose to investigate the chemistry of
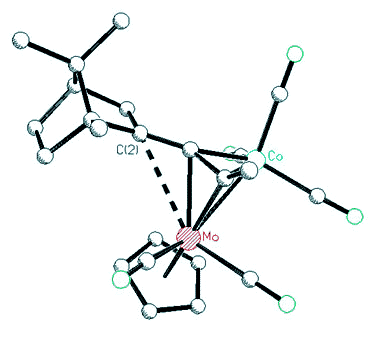
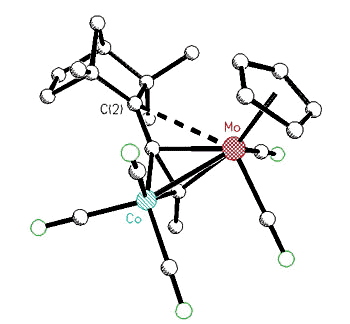
organometallic derivatives of the bornyl and fenchyl systems. While cobalt-stabilized
cations can still undergo skeletal rearrangements, incorporation of (C5H5)Mo(CO)2
vertices into tetrahedral clusters led to stable, crystalline salts containing the
2-bornyl or 2-fenchyl cations [125,138].
 |
 |
| X-ray
crystal structure of a Co-Mo alkyne cluster possessing a molybdenum-stabilized
endo-2-bornyl cation |
X-ray crystal structure of a Co-Mo
alkyne cluster possessing a molybdenum-stabilized exo-2-fenchyl cation
|
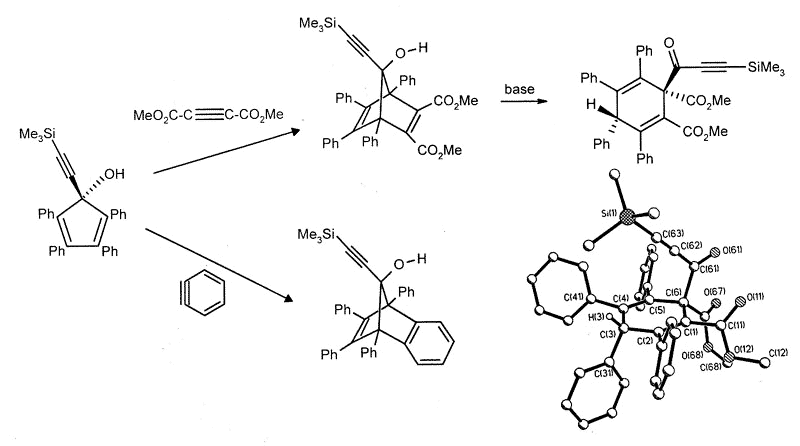
Current work is directed towards the
synthesis of metal-stabilized 7-bornyl cations via Diels-Alder additions to
alkynylcyclopentadienols, which occasionally yield surprising results. Thus, the
reaction with benzyne is normal, but the Diels-Alder adduct with dimethyl
acetylenedicarboxylate undergoes a multi-step skeletal rearrangement in the presence of
traces of base [173].