| McMaster University - Chem2O06 Lab Manual | 1997/98 |
Expt. 1, Part B. Recrystallization and Melting Point Determinations.
Introduction
1. Recrystallization
Products from an organic reaction are seldom obtained in a pure state directly from the reaction mixture. If the product is a solid, it may be purified by recrystallization from a suitable solvent. The process of crystallization depends on the fact that most organic compounds are more soluble in hot solvents than in cold, and that the impurities present will have different solubilities from the desired compounds. The procedure involves:
(a) dissolving the impure material in a minimum amount of hot solvent (if too much solvent is added, the excess can be boiled off),
(b) filtering the hot solution at its boiling point to remove insoluble impurities when present (this filtration is required only when insoluble impurities are observed),
(c) allowing the solution to cool and deposit crystals of the compound,
(d) filtering the crystals from the solution (called the mother liquor,
(e) washing the crystals with a little cold solvent to remove the mother liquor, and
(f) drying the crystals to remove the last traces of solvent.
In this experiment, recrystallization from a single solvent (water) will be illustrated.
Properties of an Ideal Recrystallizing Solvent
If recrystallization is to be effective, the solvent must be properly selected. A good recrystallization solvent should:
(a) dissolve a moderate quantity of the substance to be purified at an elevated temperature, but only a small quantity at low temperatures
(b) not react with the substances to be purified
(c) dissolve impurities readily at a low temperature or not dissolve them at all
(d) be readily removable from the purified product.
This last requirement usually means that the solvent must have a relatively low boiling point and evaporate readily.
How to Find a Good Recrystallizing Solvent
Let us assume that you have an unknown compound and you want to purify it by crystallization. First, determine its solubility properties in simple available solvents.
It is usual to try water (seldom successful), methanol, ethanol, acetic acid, petroleum ether (available in several boiling ranges) and, less commonly, ether, chloroform, ethyl acetate, acetonitrile, benzene and dimethylformamide. Add about 20 mg of material to 1 ml of cold solvent. Shake up to 3 min to help dissolution. If insoluble, warm the solvent to boiling. Note the solubility properties in each solvent (cold and hot). Ideally your material should be insoluble in cold solvent and soluble in hot solvent; if you find a solvent with these properties you can stop here as this will probably be a suitable solvent for recrystallization.
If no single solvent is found suitable, then a mixed solvent recrystallization is in order. For this you require two miscible solvents. For mixed solvent recrystallization your material should be relatively soluble in one solvent and relatively insoluble in another solvent.
For example, a substance which is very soluble in alcohol and almost insoluble in water may crystallize well from a mixture. The correct procedure is to dissolve the solid in the minimum amount of boiling alcohol and add warm water dropwise. Each drop will produce a cloudiness which at first clears on mixing. When the warmed solution just fails to clear on shaking, a few drops of alcohol are added, the mixture is re-heated and set aside for crystallization. Any insoluble impurities are best removed by filtering the hot alcohol solution before adding water. All these organic solvents have their individual hazards and contact with both liquid and vapour should be avoided. Special attention is drawn to benzene which is now recognized as a cumulative poison affecting the blood. Even low concentrations of benzene vapour should therefore be avoided.
Some Further Points on Recrystallization
Often, information is available concerning solvents suitable for recrystallizing a particular compound. If it is not, several solvents may be tested. This is done by placing a small amount of the substance to be purified in each of several test tubes and adding a small amount of a different solvent to each. Solubility is noted in the cold solvent and at its boiling point; one also observes whether abundant, well-formed crystals are produced on cooling the hot solutions.
To obtain a good recovery of purified material, the use of unnecessarily large volumes of solvent must be avoided. The amount of pure material lost by retention in the mother liquor will be minimized if the substance is dissolved in the smallest amount of hot solvent. In practice, 3-5% more than the minimum is used so that the hot solution will not be quite saturated. This helps prevent the separation of crystals and clogging of the filter paper during filtration of the hot solution.
Traces of coloured matter or resinous impurities may sometimes be removed with selective adsorbents, such as finely divided charcoal (Norit, Darco). A small amount of decolourizing charcoal is added to the hot solution before it is filtered. Excess of decolourizing agent should be avoided, however, because it may also adsorb appreciable amounts of the substance being purified.
Some substances readily form supersaturated solutions, and crystallization may not occur spontaneously when the hot solution is cooled. In such situations, crystallization can sometimes be initiated by scratching the walls of the vessel with a stirring rod, beneath the surface of the solution. The best method to induce crystallization is to "seed" the cold solution with one or two pure crystals of the substance being purified. Although some compounds crystallize readily, others may separate from solution as oils, and may require hours or even days before they crystallize.
Apparatus for Vacuum Filtration
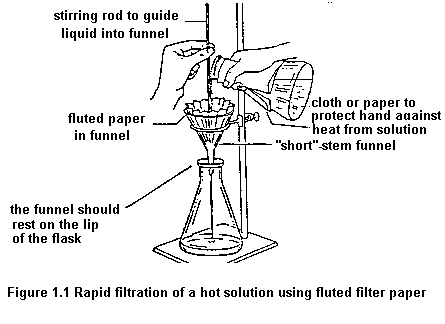
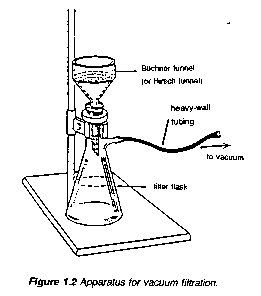
To remove insoluble impurities and decolourizing charcoal, hot solutions must be filtered rapidly. Otherwise, the solution cools and crystals deposit prematurely. Rapid filtration may be accomplished by using fluted filter paper (paper folded with many pleats to give a large surface, Figure 1.1) or, more commonly, by using a vacuum to increase the filtration rate. The apparatus for vacuum filtration is shown in Figure 1.2. A Buchner or a Hirsch funnel is fitted to a filter flask with a neoprene adaptor. A disk of filter paper of a size adequate just to cover all the holes in the funnel is placed in the funnel and moistened with some of the solvent used in the recrystallization. The filter flask is then joined to the aspirator and vacuum is applied. When the filter paper is drawn down tightly to the funnel, filtration of the solution is begun. During filtration, it is important not to turn off the aspirator or to decrease the aspirator flow rate as this may cause "suck back" of tap water into the filtration flask. For this reason, it is also important to break vacuum at either hose connection before turning the aspirator off.
Melting Point Determination
A melting point can be used to identify a substance and to get an indication of its purity. The melting point (or freezing point) of a solid is the temperature at which the solid exists in equilibrium with its liquid state under an external pressure of one atmosphere. More precisely, it is the temperature at which the vapor pressure of the solid phase becomes equal to the vapor pressure of the liquid phase. Experimentally, it is extremely difficult to establish the exact temperature at which this equilibrium is established; therefore, the temperature range over which liquid and solid are found to coexist is called the melting point. For example, a solid may be reported to have a `melting point' of 100-101oC; this means that, on heating slowly, the first droplet of liquid was observed at 100oC and the last crystal of solid disappeared at 101oC.
Both the melting point range (the interval between the beginning of liquefaction and complete liquefaction) and the temperature of complete liquefaction are valuable indicators of the purity of the solid compound. A pure crystalline organic compound usually possesses a sharp melting point and it melts completely over a narrow temperature range of not more than 0.5-1.0oC, provided good technique is followed. The presence of even small amounts of impurities usually produces a depression of the temperature at which melting is complete and usually produces a marked increase in the width of the melting point range. For example, if a sharp-melting unknown substance X is suspected of being identical with some known substance A, the two should have the same melting points. If A is reported to have a melting point rather different from that observed for X, the two substances may be identical (the small differences being due to variations in technique of determining the melting points). Whether they are indeed identical can often be deduced quickly if a sample of A is available, by determining a mixture melting point. A mixture of X and A should have the same melting point as that of either substance alone, provided the two substances are identical.
If X and A are not the same substance (even though they separately have the same melting point), then a mixture of the two will usually show a lower melting point and a broader melting point range than either substance alone. This is because each substance acts as an impurity in the other. Miscible or partially miscible impurities, even when present in small amounts, usually lower the melting point and broaden its range.
A wide melting point range usually indicates that a substance is impure, but it may also result from the fact that the pure substance undergoes some decomposition prior to reaching its melting point. In some cases, the material undergoes a slight liquefaction and contraction at a temperature below the true melting point; in others, the material may decompose and discolour so badly that a definite melting point cannot be observed.
Theory of Melting Point Depression
The theory underlying the solid/liquid change of state is based primarily on the phenomenon of vapor pressure. Both liquid and solid forms of a compound exert vapor pressure; vapor pressure increases with increasing temperature but that of the solid increases more rapidly than that of the liquid. At the melting point, the vapour pressures of the solid and liquid phases are equal. A soluble impurity lowers the partial vapour pressure of the pure substance in the melt and thus lowers the temperature necessary for melting. The temperature dependence of the vapor pressure of a pure compound, A, is concisely summarized by a typical phase diagram, Figure 1.3.
Two points in Figure 1.4 may be identified: TB, the normal boiling point, the temperature at which the vapor pressure of the liquid equals atmospheric pressure, and TM, the melting (or freezing) point, the point on the solid liquid line equal to the atmospheric pressure.
The "true" melting point is the temperature at which all three phases (vapor, liquid, solid) coexist -- i.e., the "escaping tendencies" from any one phase to another are equal. The total pressure of the system is, then, the vapor pressure of the compound at that temperature.
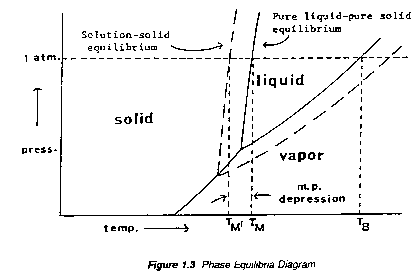
"Ordinary" melting points, however, are determined at atmospheric pressure and differ slightly from true melting points. Melting points are not markedly affected by modest changes in external pressure because the solid/liquid phase transition does not involve a significant change in molal volume. However, at very high pressure, startling differences may be observed: e.g., at 33,880 atm., the melting point of water is 166.6oC instead of 0oC.
The effect of a small amount of an impurity, B, on the melting point of A may be evaluated by the following considerations. Suppose that a small amount of B were introduced into the equilibrium mixture of pure, solid and liquid A and that B immediately dissolves in liquid A but not in solid A, which it cannot quot;penetrate".
According to Dalton's Law the vapor pressure of a liquid solution is the sum of the partial pressures of the components. Therefore, the presence of this dissolved impurity will lower the partial vapor pressure due to liquid A throughout the whole temperature range as shown by the `dashed' curve in Figure 1.3. It is clear then that the vapor pressure of solid A will become equal to that of liquid A in solution at temperature TM' which is below the melting point TM of pure A -- i.e. the effect of the impurity is to depress the melting point of the pure compound.
Further additions of small quantities of the impurity will produce corresponding lowerings in the partial vapor pressure of A in the liquid melt and hence also, in the melting point of compound A. Finally, however, a limiting point is reached at which the impurity concentration is just sufficient to `saturate' liquid A; any additional impurity does not dissolve and cannot further depress the melting point. This point is known as the Eutectic Point and the limiting temperature is called the Eutectic Temperature and the composition of the melt, the Eutectic Composition. Alternatively, the eutectic temperature can be described as the temperature below which a mixture of A and B cannot exist as a liquid; or, the temperature at which A and B can co-crystallize from the liquid melt.
Eutectic Point
The nature of the eutectic point and, more importantly, its influence on the observed melting point range are more effectively illustrated by a generalized, equilibrium temperature versus composition diagram, Figure 1.4. In this diagram, point a represents the melting point of pure compound A; the curve aE represents the temperatures at which solutions of different concentrations of B (the impurity) in A are in equilibrium with solid A -- i.e., for any particular concentration, the curve indicates the temperature at which the last trace of A will melt when the solid mixture is heated or the temperature at which the first trace of A will crystallize if the liquid mixture is cooled. Similarly, point b represents the melting point of pure B and the curve bE represents the temperatures at which solutions of different concentration of A (the impurity) in B are in equilibrium with solid B. At the eutectic point, E, both solid components can exist in equilibrium with a liquid solution of that particular composition. One might consider the liquid at the eutectic point to be a saturated solution of either solute A in solvent B or, solute B in solvent A. Cooling of the eutectic liquid will bring about crystallization (freezing) of both A and B at a constant temperature, the eutectic temperature and at a constant composition, the eutectic composition.
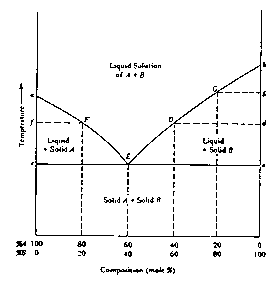
To evaluate the effect of an impurity on the melting point range, consider the effect of heating a mixture of A (80%) with an amount of impurity, B (20%), less than the eutectic concentration. To establish the range we must know where the mixture will begin to melt. When heat is applied, the temperature of the solid mixture will rise; no changes in the physical state of the system will occur until the eutectic temperature is reached. At that temperature, assuming that the heating can be controlled sufficiently that equilibrium conditions are ensured, appropriate numbers of molecules of A and B already in contact at the crystal/crystal interfaces will `melt' to form a liquid phase of eutectic composition (in this case 60/40). As heating is continued more A and B will melt at the eutectic composition and the eutetic temperature, until all of B (the minor component) is entirely melted leaving only solid A in equilibrium with the eutectic liquid. On further heating, the remaining solid A will begin to melt. This, however, will raise the percentage of A in the liquid above the eutectic composition. Since the partial vapor pressure due to A in the liquid is thereby increased, the temperature (melting point.) at which solid A is in equilibrium with the liquid must also rise. In this fashion, melting will continue, at progressively increasing temperatures (represented by the curve EF in Figure 1.4) until the last trace of solid A becomes liquid at temperature F. Hence, if perfect equilibrium conditions were maintained, the melting point range for such a mixture would be from E, the eutectic temperature, to F.
Similarly, if we were to consider a solid mixture with a composition to the right of point E in Figure 1.4, we would speak of a compound B with impurity A and the rising temperature during the melting process would follow curves E D or E G.
In theory, then, for any solid compound containing a relatively small amount of impurity, melting will begin at the eutectic temperature and be complete at some temperature lower than the melting point of the pure compound. Moreover, if the concentration of the impurity were increased, the upper limit of the melting would be lowered and therefore the melting range decreased.
In practice, however, equilibrium conditions are almost never achieved and, moreover, it is extremely difficult to detect the initial melting or eutectic condition. If only a very small amount of impurity is present (which is most often the case) the temperature may rise several degrees above the eutectic temperature before sufficient liquid phase accumulates to be visible to the human eye. Nevertheless, the temperature at which the last crystal disappears can be determined quite accurately.
Therefore, as actually observed, a nearly pure solid compound tends to show a narrow melting range with an upper limit near the true melting point whereas a rather impure compound usually shows a broader melting range with the maximum temperature considerably below the true melting point.
It is clear from further consideration of Figure 1.4 that the effect of an impurity is not directly dependent on its own melting point. Thus, for example, a solid impurity whose melting point is higher than that of the compound will still depress the latter's melting point (at least in amounts less than that of the eutectic composition). It should also be noted that a sample whose composition is exactly that of the eutectic mixture will exhibit a sharp melting point, melting completely at the eutectic temperature. Thus a eutectic mixture may sometimes be mistaken for a pure compound. Such infrequent occurrences may be identified by adding a small amount of either component (assuming one knows what the components are) and observing that the melting point rises. Finally, the eutectic point is not limited to mixture compositions around 50/50 (60/40 in the arbitrary example shown in Figure 1.4) but may in principle occur at any composition depending only on the specific properties of the particular components.
General Technique for Melting Point Determination>
To determine the melting point of a crystalline substance, a small amount of the finely powdered crystals is introduced into a thin-walled capillary tube; the latter is placed on an electrically heated "hot-stage" and heated. Two temperatures are recorded, the temperature at which the substance begins to liquefy and that at which it becomes completely liquefied. The observed melting point range is the interval between these two temperatures.
A pure crystalline organic substance usually possesses a sharp melting point; that is, it melts completely over a very short temperature range, usually not more than 0.5-1.0oC, provided good experimental technique is used. The melting point range is influenced not only by the purity of the material but also by the size of the crystals, the amount of material, the density of its packing in the tube, and the rate of heating the hot stage. A finite time is required to transfer heat from the hot-stage both through the walls of the capillary tube and throughout the mass of the sample. If the hot-stage is heated too quickly, its temperature will rise several degrees during the time lag required for the melting process to occur; this results in an observed range higher than the true one.
To obtain good results it is essential, in the vicinity of the melting point, to heat the melting point hot-stage slowly at a uniform rate, about 2 degrees per minute. To minimize lag in the melting process and heat transfer, a small amount of material, finely powdered and densely packed in a thin-walled capillary of small diameter, should be used. The height of solid in the capillary tube should be just enough to permit good observation of the behaviour on melting (about 3-4 mm).
The behaviour of a material on melting should be observed and recorded carefully, as, for example: melts sharply at 89.0-89.5oC; or mp 131-133oC, with decomposition; or discolours at 65oC, melts slowly at 67-69oC.
The Melting Point Stage
Several types of apparatus used to determine melting points are shown in Figure 1.5. Types A to C are older versions and use liquids as the heat transfer agents. In this laboratory you will use apparatus D which comes in two versions: one for use with a capillary tube and the other for use with a microscope cover slide. You should use whatever version is available to you in your labaratory room. Make sure that you use a high temperature (250 or 360oC) thermometer!
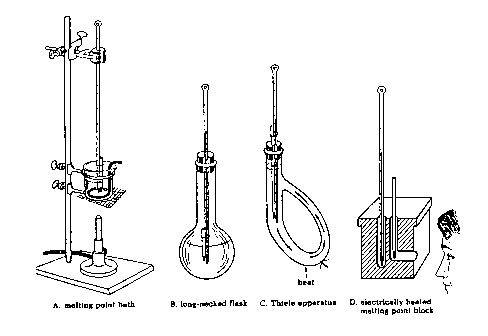
Determination of Melting Points
Procedure: Fill a melting point tube with the sample of interest by thrusting the open end into the powder several times. In order to work the plug of solid material down to the sealed end of the tube, vigorously tap the sealed end on the table or lightly draw a file across the tube held loosely in the hand. Repeat the procedure until the tube contains a 3 mm column of densely packed powder in the bottom. Place the capillary in the melting point stage and turn on the power and allow the hot-stage temperature to rise fairly rapidly to within 15-20oC below the expected melting point of the compound. However, during the determination of the actual melting point range, the temperature should not rise more rapidly than 2 or 3o per minute. Therefore, decrease the rate of heating about 15o below the expected melting point. Observe and record the melting range.
[Note: frequently it is faster to prepare two capillaries, perform one rapid estimate of your melting point, and then do a more careful one using a 2-3oC temperature rise per minute near your "estimate".]
Questions
1. What effects do impurities have on the melting point of an organic compound?
2. How could the identity of a compound be established by melting point determination?
References:
Pasto and Johnson, Organic Structure Determination, pp. 57-65.
Adamson, Textbook of Physical Chemistry, pp. 395-396.
CRC Press, Handbook of tables for Organic Compound Identification.
| Go to: | Instructions for Printing this Document Experiment 1 Part B Procedures Chem2O06 Home Page. |
04sep97; wjl